Statistiche di rischio

Carcinoma del colon ereditario non poliposico
(HNPCC - Hereditary Non-Polyposys Colon Cancer)
L’HNPCC (Hereditary Non-Polyposis Colon Cancer - HNPCC),
conosciuta anche come Sindrome di Lynch, è una
malattia tumorale autosomica dominante
caratterizzata da due manifestazioni fenotipiche:
La
sindrome di Lynch I,
che è caratterizzata
dall'insorgenza di una neoplasia al colon ad un'età media di circa
45 anni.
La sindrome di Lynch II,
che
oltre al tumore al colon comprende lo sviluppo di neoplasie
extracoloniche, a livello dell'endometrio, dell'ovaio, dello
stomaco, del tratto urinario, dei dotti biliari.
L'HNPCC è causata da un evento
mutazionale ricorrente a livello di uno dei 4 geni attualmente
conosciuti essere coinvolti nel controllo e nella riparazione
degli errori di replicazione del DNA in tutte le cellule del
corpo. Circa il 90% delle mutazioni avvengono a livello dei
geni
MSH2 e MLH1 (60% in MSH2 e 30% in MLH1) mentre solo raramente
coinvolgono i geni PMS1 e PMS2. Quando avviene un evento
mutazionale a livello di uno di questi geni, la capacità di
effettuare la riparazione degli errori intercorsi durante la
duplicazione del DNA diminuisce, e di conseguenza le mutazioni
iniziano ad accumularsi nella cellula, conducendo allo sviluppo
neoplastico.
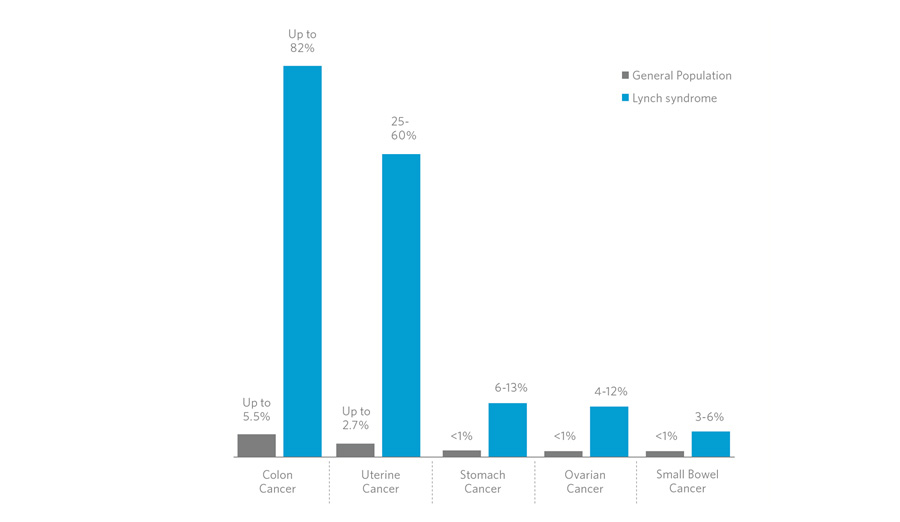
Nella popolazione
generale, il rischio di sviluppare un tumore al colon nel corso
della vita è attualmente stimato intorno al 6%. Per quei pazienti
(sia uomini che donne) che sono portatori di mutazioni HNPCC, il
rischio di sviluppare il tumore al colon è stimato intorno al
75-90%.
Le neoplasie extracoloniche rappresentano
un'importante complicazione, la più comune delle quali è
rappresentata dal tumore all'endometrio, il cui rischio
complessivo è stato valutato intorno al 30%, contro il 3% della
popolazione generale. Il rischio di sviluppare un tumore ovarico è
3.5 volte più alto e può ricorrere 20 anni prima rispetto alla
popolazione di riferimento.
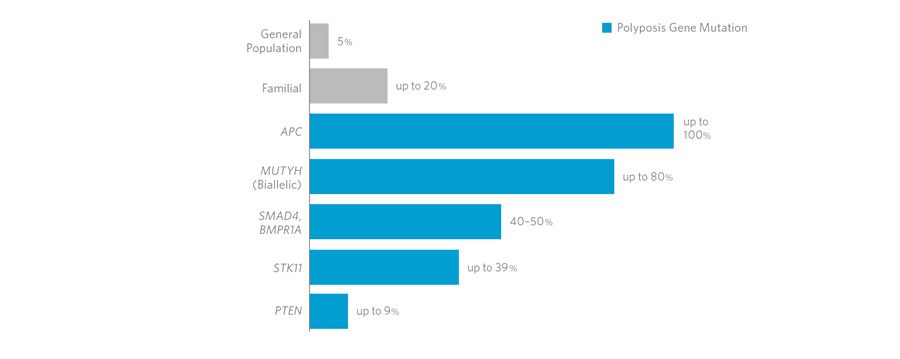
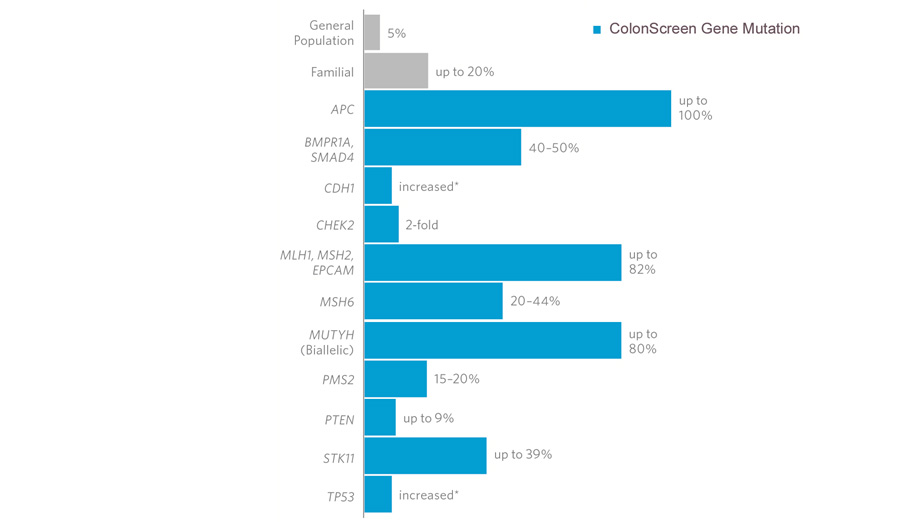
La Poliposi Adenomatosa Familiare (FAP
o Adenomatosis coli o Poliposi Familiare del colon-retto)
è una sindrome relativamente rara caratterizzata dalla comparsa,
di solito in età giovanile, di centinaia o migliaia di adenomi
distribuiti nei vari segmenti del grosso intestino. Se non
trattata, la FAP progredisce quasi invariabilmente verso lo
sviluppo di uno o più carcinomi colorettali, di solito nella terza
o quarta decade di vita; la comparsa di lesioni maligne può essere
prevenuta attraverso un’attenta sorveglianza endoscopica ed un
tempestivo intervento chirurgico. La FAP rappresenta pertanto una
condizione precancerosa “obbligata”, nel senso che l’individuo
affetto e non trattato in tempo va incontro al manifestarsi di un
carcinoma invasivo con certezza pressoché assoluta.
La FAP
è una malattia ereditaria, autosomica dominante, solitamente ad
elevata penetranza; ne consegue che un’attenta analisi dell’albero
genealogico può permettere di individuare in ogni fratria i
soggetti a rischio, nei quali iniziare una accurata sorveglianza
endoscopica. Oggi ciò è reso ancor più agevole grazie
all’identificazione del gene responsabile, quando mutato, della
malattia (gene APC, Adenomatous Polyposis Coli).
In un
sottotipo di pazienti, una mutazione nel gene MUTYH (1p34.1) causa
una poliposi autosomica recessiva, la poliposi adenomatosa
familiare legata a MUTYH, caratterizzata da un leggero aumento del
rischio di sviluppare tumori colon-rettali e polipi/adenomi nel
tratto gastrointestinale superiore e inferiore.