Risk Tables

HNPCC - Hereditary Non-Polyposys Colon Cancer
The HNPCC (Hereditary Non-Polyposis Colon Cancer - HNPCC),
also known as Lynch Syndrome, is an autosomal,
dominant cancer with two phenotypes:
Lynch
Syndrome I,
with the onset of a colon
neoplasm at around 45 years of age.
Lynch
Syndrome II,
which, in addition to colon cancer,
includes the development of extracolonic neoplasms,
at an endometrial, ovarian, urinary tract and biliary duct level.
The HNPCC is caused by a recurring mutation in one of the 4 genes
that are currently known to be involved in the check and repair of
DNA replication errors in all the cells of the body. Around
90% of mutations occur in MSH2
and MLH1 genes (60% in MSH2 and 30% in MLH1) and
only rarely involve PMS1 and PMS2 genes. When a mutation occurs in
one of these genes, the ability to repair errors during DNA
duplication decreases and, as a consequence, mutations accumulate
in the cells, leading to a neoplasm.
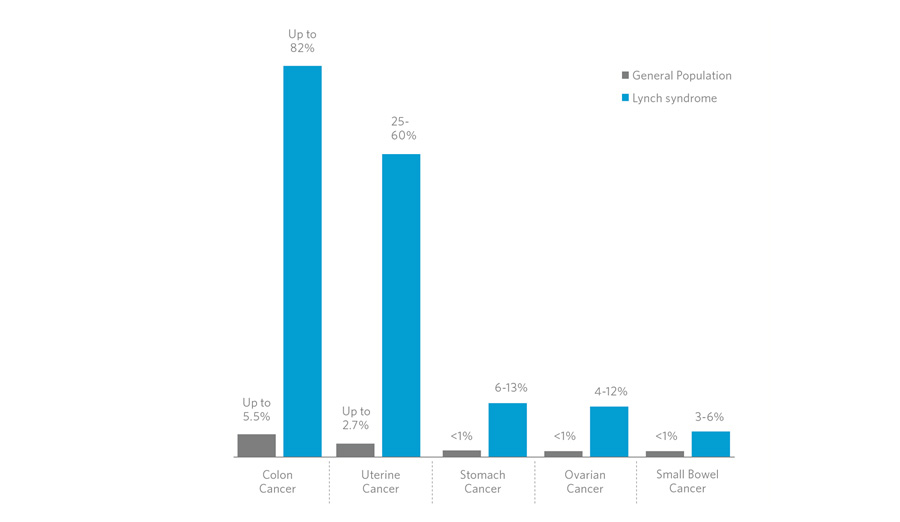
According to statistics, the risk for the general
population to develop colon cancer in
life is around 6%. For patients (both male and
female) carrying HNPCC mutations, the risk of
developing colon cancer is around 75-90%.
Extracolonic neoplasms are a severe complication, the most common
of which is endometrial cancer. The overall risk
of this type of cancer is around 30%, compared to 3% among the
general population. The risk of developing
ovarian cancer is 3.5 times higher and it may
occur 20 years earlier compared to the reference population.
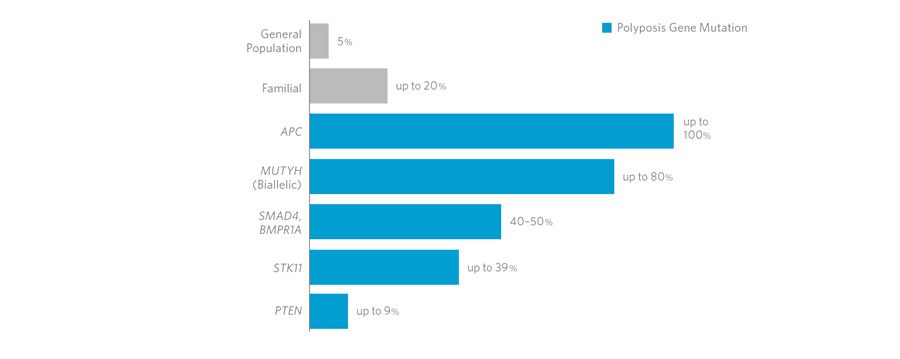
The Familial Adenomatous Polyposis (FAP or
Adenomatosis coli or Colorectal Familiar Polyposis) is a
relatively rare syndrome that occurs, usually at an early age,
with the development of hundreds to thousands of adenomas on the
large intestine. Without treatment, FAP progresses almost
invariably with one or more colorectal carcinomas, usually when
the individual is 30 to 50 years old; the onset of malignant
lesions may be seen with careful endoscopic checks and prompt
surgical intervention. The FAP is a "compulsory" precancerous
condition, because the person that is affected and not treated
will almost certainly develop invasive carcinoma.
The FAP is a
hereditary, autosomal, dominant disorder, usually at high
penetration. Therefore, a careful analysis of the family history
could help detect subjects at risk in each phratry on which
accurate endoscopic checks must be carried out. Presently, this
process is easier, thanks to the detection of the gene that, once
mutated, generates the disorder (APC gene, Adenomatous Polyposis
Coli).
In a sub-type of patients, a mutation of the MUTYH
(1p34.1) gene leads to an automosal recessive polyposis, the
familial adenomatous polyposis linked to the MUTYH, which is
characterised by a small increase in the risk of developing
colorectal cancer and polyps/adenomas in the higher and lower
gastrointestinal tract.